edgeR で発現変動遺伝子を検出する
次世代シークエンスで遺伝子の発現量を調べるには、リファレンスの配列にマッピングを行い、遺伝子ごとにマッピングできたリード数を計算すれば良いですが、複数のサンプルで発現量が異なる遺伝子を検出するには、統計学的な手法が必要になります。
本記事では、R のパッケージである edgeR を使用することで、発現変動遺伝子を検出する方法を解説します。
edgeR とは何か
-- ChatGPT --
edgeR(エッジアール)は、RNA-Seq などのカウントデータから差次的発現遺伝子(DEGs)を統計的に見つけるための R パッケージです。
🔬 edgeR でできること
・RNA-Seq の raw カウントデータを扱える
・2群または多群の差次的発現解析(DEG解析)
・ノイズ除去や正規化(ライブラリサイズ補正)
・複雑な実験デザイン(多因子解析)にも対応
⚙️ 背景にある統計モデル
・edgeR は 「負の二項分布(Negative Binomial)」 に基づいて発現量のばらつきをモデル化しています。
・これにより、少ないサンプル数でも信頼性のある解析が可能です。
(以下略)
----
edgeR を使用することで、統計学的な手法に基づいた発現変動遺伝子 (DEG) の検出を自動で行うことができます。
edgeR で DEG を検出する
それでは、実際に edgeR を使用して DEG を検出していきます。今回は、以下のような6サンプルのリードカウントデータを使用します。このフォーマットのファイルを作る方法については、こちらの Salmon の記事をご覧ください。
注意する点として、今回使うのはリードカウントのデータです。edgeR は実行中にリードカウントから正規化を行うので、TPM を使用すると正規化を二重にすることになり、正しい結果が得られなくなります。
# head でファイルの先頭を表示
head count.tsv
# 実行結果
Name sample1 sample2 sample3 sample4 sample5 sample6
gene_1 102 174 149 800 151 257
gene_2 1 4 0 4 1 1
gene_3 104 105 61 0 1 1
gene_4 74 52 90 24 4 10
gene_5 153 134 176 236 76 67
gene_6 0 4 3 1060 212 183
gene_7 3 7 10 84 21 22
gene_8 48 55 53 96 24 13
gene_9 48 28 31 16 0 1以上のような、「count.tsv」ファイルを入力とします。中には6サンプルのリードカウントが含まれていて、最初の3つと後ろの3つが、それぞれ同じ条件のバイオロジカル・リプリケイトです。最低2回の複製がないとエラーになります。
それでは、edgeR を使用していきます。最初に R をインストールして、その後 edgeR をインストールすると、R 内で edgeR が使用できるようになります。インストールの方法については自分の環境などに合わせて自由に行うことができますが、私の場合は Docker を使用しました。Dockerfile をカスタマイズすることで、R と edgeR を一括で導入したコンテナを作成できます。
M4 Mac で Docker を使う場合は、以下のような Dockerfile を用意して、ビルドします (Dockerfile を作りたい場合は、ChatGPT に頼めば自分の環境に合わせて作ってくれます)。
FROM ubuntu:22.04
# タイムゾーン設定
ENV TZ=Asia/Tokyo
RUN ln -fs /usr/share/zoneinfo/$TZ /etc/localtime && \
DEBIAN_FRONTEND=noninteractive apt-get update && \
apt-get install -y tzdata
# パッケージインストール
RUN apt-get update && apt-get install -y \
sudo \
wget \
vim \
gnupg \
software-properties-common \
ca-certificates && \
mkdir -p /etc/apt/keyrings && \
wget -qO- https://cloud.r-project.org/bin/linux/ubuntu/marutter_pubkey.asc | gpg --dearmor > /etc/apt/keyrings/cran-archive-keyring.gpg && \
echo "deb [signed-by=/etc/apt/keyrings/cran-archive-keyring.gpg] https://cloud.r-project.org/bin/linux/ubuntu jammy-cran40/" > /etc/apt/sources.list.d/cran.list && \
apt-get update && \
apt-get install -y r-base \
libcurl4-openssl-dev \
libssl-dev \
libxml2-dev \
&& apt-get clean
# R パッケージ edgeR のインストール
RUN Rscript -e "install.packages('BiocManager', repos='https://cloud.r-project.org')" && \
Rscript -e "BiocManager::install('edgeR')"
# 作業ディレクトリを設定
WORKDIR /work
# デフォルトで bash を起動
CMD ["bash"]# ビルド
docker build -t ubuntu-r .
# run
docker run -it --rm -v "$(pwd)":/work ubuntu-rR と edgeR のインストールができたら、R を開いて以下のコマンドを順番に実行してください。
サンプル数などの条件が異なる場合は、自分の条件に合わせてコマンドを調整してください。
## edgeR を使えるようにする
library("edgeR")
## DEG 検出
# 入力ファイル名を調整 ("count.tsv"のところ)
count_data = as.matrix(read.table("count.tsv", sep = "\t", header = T, row.names = 1))
# サンプル数を調整 (c の中、私の場合 W が3反復と M が3反復)
sample_condition = factor(c("W", "W", "W", "M", "M", "M"))
deg_list = DGEList(counts = count_data, group = sample_condition)
deg_list = calcNormFactors(deg_list)
deg_list = estimateCommonDisp(deg_list)
deg_list = estimateTagwiseDisp(deg_list)
exact_test_result = exactTest(deg_list)
output_table = as.data.frame(topTags(exact_test_result, n = nrow(count_data)))
write.table(output_table, "edgeR_result.txt", sep = "\t", quote = F)
## MA-plot をかく
point_colors <- ifelse(output_table$FDR <= 0.05, "red", "black")
png("MA_plot.png", width = 1000, height = 800)
plot(output_table$logCPM, output_table$logFC,
col = point_colors, pch = 20,
xlab = "A-value", ylab = "M-value",
main = "MA-plot")
dev.off()実行できたら、「edgeR_result.txt」と「MA_plot.png」が出力されます。とりあえず「edgeR_result.txt」の方を見てみます。
# R を終了する (R の変更を保存するか聞かれますが、どちらでも良いです)
q()
n
head edgeR_result.txt
# 実行結果
logFC logCPM PValue FDR
gene_293 -11.0041839250789 8.40740208112218 7.5188230223447e-50 7.5188230223447e-46
gene_44 -10.9696660983025 11.518575357395 5.59089041946381e-37 2.7954452097319e-33
gene_296 5.13908452536243 8.12693586872543 5.32628839594311e-28 1.77542946531437e-24
gene_90 -5.89535916838515 7.56791133343261 4.27196289510595e-26 1.06799072377649e-22
gene_96 11.852005531074 7.44324004034648 5.92084590760058e-25 1.18416918152012e-21
gene_284 -6.5601289027958 5.95353988245981 1.3206945827815e-24 2.20115763796916e-21
gene_20 10.0158208145008 5.64011711052224 4.89129721243379e-24 6.98756744633399e-21
gene_290 -6.49088707304741 6.12660851573131 1.27541138793973e-23 1.59426423492466e-20
gene_80 -8.76519924814903 6.95980105380202 1.96728203600726e-23 2.08592843945838e-201列目から順番に、遺伝子名、発現量の比 (M-value)、発現量の平均 (A-value)、検出結果 (P-value)、多重検定補正の結果 (FDR) なのですが、1行目に Name がなくて、列が一つずれています。R では行の名前を含む列のヘッダーが省略されるので、Name の表示がなくなってしまっているのです (上の count_data = as... の後に「head(count_data)」を実行すると Name が消えているのがわかります)。
そのため、以下のコマンドで1行目を編集しておきましょう。このコマンドは、Mac か Linux であれば使えるはずです。
# logFC を 「Name logFC」(タブ区切り) に変更して tsv で保存
sed -e "s/logFC/Name\tlogFC/g" edgeR_result.txt > edgeR_result.tsv
# 確認
head edgeR_result.tsv
Name logFC logCPM PValue FDR
gene_293 -11.0041839250789 8.40740208112218 7.5188230223447e-50 7.5188230223447e-46
gene_44 -10.9696660983025 11.518575357395 5.59089041946381e-37 2.7954452097319e-33
gene_296 5.13908452536243 8.12693586872543 5.32628839594311e-28 1.77542946531437e-24
gene_90 -5.89535916838515 7.56791133343261 4.27196289510595e-26 1.06799072377649e-22
gene_96 11.852005531074 7.44324004034648 5.92084590760058e-25 1.18416918152012e-21
gene_284 -6.5601289027958 5.95353988245981 1.3206945827815e-24 2.20115763796916e-21
gene_20 10.0158208145008 5.64011711052224 4.89129721243379e-24 6.98756744633399e-21
gene_290 -6.49088707304741 6.12660851573131 1.27541138793973e-23 1.59426423492466e-20
gene_80 -8.76519924814903 6.95980105380202 1.96728203600726e-23 2.08592843945838e-20これで列がしっかり整った tsv ファイルを作成することができました。コマンドを使わなくても、普通にファイルの先頭に Name とタブを追加するだけでも大丈夫です。
次に、「MA_plot.png」を開いてください。黒点と赤点が描かれた散布図になっているはずです (DEG がない場合、黒点だけになります)。先ほど軽く書きましたが、M-value はリードカウントを正規化した値の比 (上の R のコマンドの中の「M」と「W」に指定したサンプルで比較しています) で、A-value はリードカウントを正規化した値の平均値です (どちらも log2 をとっています)。
そのため、M-value の絶対値が大きいほど、その遺伝子の発現量が大きく異なっていることを表していて、A-value が大きいほど、その遺伝子の発現量がサンプル全体で大きいことを表しています (あくまで平均ですが)。
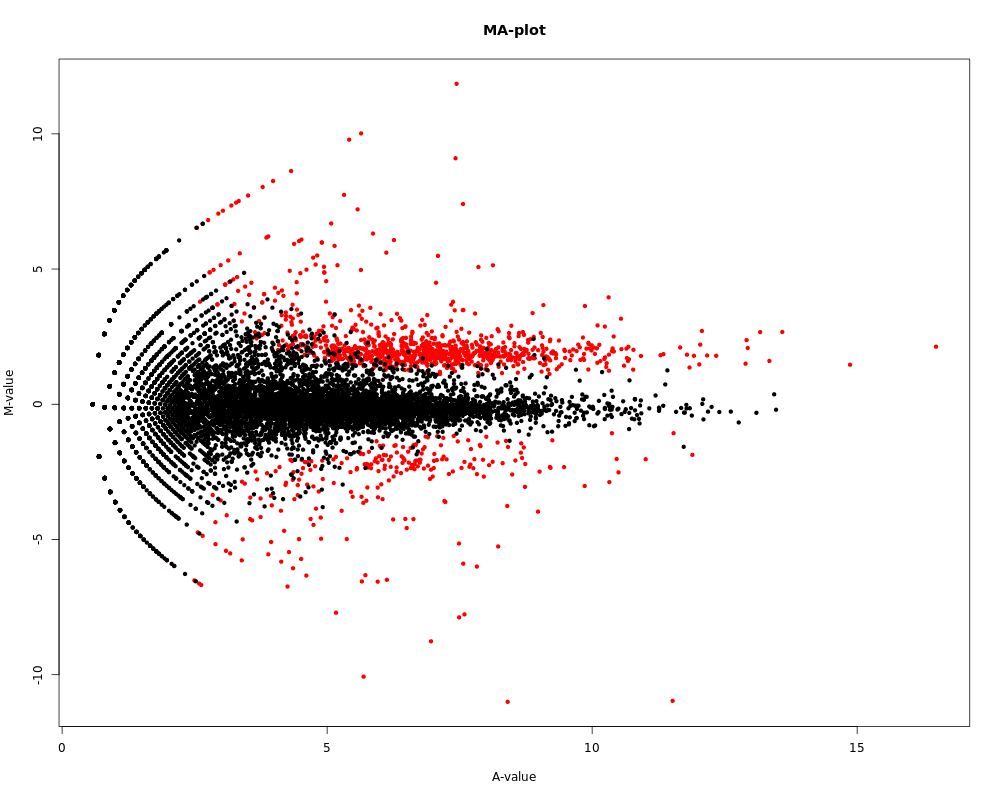
今回の場合、FDR が0.05以下の点を赤色にしているので、それが発現変動遺伝子です。
DEG を検出できた後は、以前解説した Blast による相同性検索や、 GO エンリッチメント解析などを行うことで、それらの遺伝子がどのような役割を持っているのかを調べることができます。
今回は以上です。
おまけ: 書籍紹介
バイオインフォマティクスの勉強に役立つ本をいくつか紹介します。
リンクをクリックすると、Amazon のページに飛びます。
・バイオインフォマティクス入門
生物学、コンピュータ、バイオインフォマティクスについて、基礎的な知識を習得するのに役立つ本です。見開きのページごとで内容がまとめられているので、毎日少しずつ勉強したいという方にもおすすめです。
・RNA-Seqデータ解析 WETラボのための超鉄板レシピ
具体的な解析方法が載っている本です。
私が使用しているパソコンはこちらです。環境構築などを同じ条件でやりたい方は、よろしければどうぞ。